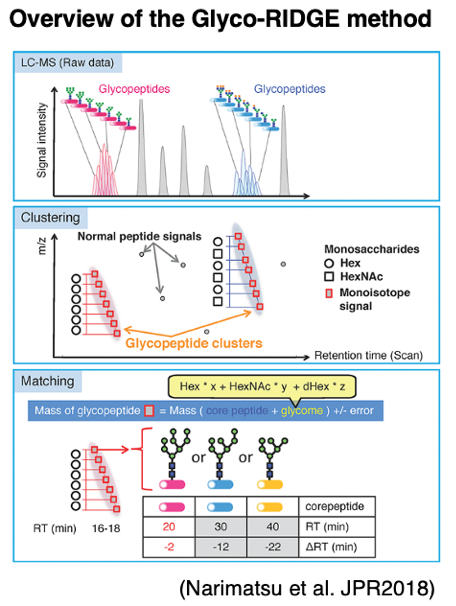
GRable is a software to estimate site-specific glycan compositions of glycopeptides, using an MS1-based glycoproteomic method named “Glyco-RIDGE” [Narimatsu et al. JPR2018]. You can freely try this software on this site!
Please visit to [more details] including relevant references.
GRable requires following 4 files (please see [Note 1])
LC/MS data of glycopeptides (.mzML):
The LC/MS data (.raw) should be deconvoluted and exported as an mzML format using Proteome Discoverer. ([Note 2, 3])
Core peptide list (.xlsx):
This list contains information on existing core peptides identified by PNGase-mediated deglycosylation followed by LC/MS analysis. ([Note 4])
Glycan point list (.xlsx):
The list of possible glycan compositions. Points are given for each possible glycan composition. Unusual compositions considered from biosynthetic pathways can be included in the list to help select appropriate matches.
LC/MS2 data of glycopeptides (.mgf):
Peak list of MS2 spectra extracted from the raw data of 1, as mgf file. ([Note 5])
As a tutorial, example data for these four files is provided below. Download them and try to parse them. You can also analyze your own dataset by preparing four files in the same format as the example data.
Detailed information on parameter settings can be found in the manual. The default settings can be used for analysis of sample data.
Access to GRable (Notice)
Please note that uploaded data and analysis results are visible to every person who use this site. We recommend that you delete your data including the result files immediately after the trial. We do not guarantee that other users will not see your results. ([Note 6])
If you agree these notices, please click on the button and log-in using the username and password indicated below. Please note that the username and password are fixed for testing purposes.
(Username: grable, Password: grable)
Notes
All data should be less than 300 MB each.
GRable is developed using Thermo Scientific data. Therefore, data from other manufacturer’s equipment are currently not supported. Also, LC/MS using C18 column is assumed.
The analytical data require a mass accuracy better than 5 ppm. This is because GRable assigns glycopeptide signals using the accurate mass difference between glycopeptides with the same core peptide and estimates glycan composition based on the mass difference between the core peptide and the glycopeptide. If the mass accuracy is low, the correct core peptide cannot be identified, increasing the possibility of obtaining erroneous results.
This analysis should preferably be performed under the same LC conditions as the measurement of glycopeptide sample. In the case of highly purified protein, presumed peptide sequences are applicable.
By the GRable algorithm, multiple core peptides may match to single cluster. Then, it uses some surrounding information to select the most plausible match among them, e.g., sum of glycan points of cluster member, retention time difference with core peptide, signal intensity of core, etc. In addition, if a cluster member has acquired MS2, the soft will use that information.
In the future, we plan to develop an updated version that can handle data from manufacturers other than Thermo Scientific, and that allows individual user management.
If you belong to an academic research institute, you can use the full version without functional limitations by concluding a joint research agreement between your organization and AIST. Please contact us: M-GRable-inquiry-ml[at]aist.go.jp (at=@).